IRB Protocol and Consent Form Resources
Protocol Resources
IRB Consultation Services:
The IRB Consultation Service is provided to facilitate the submission of complete applications to the IRB, which will in turn reduce IRB review time; increase compliance in the conduct of human subjects research; and facilitate required reporting to the IRB.
Questions about preparing an IRB submission, responding to IRB requests, understanding IRB policies, obtaining informed consent and reporting unanticipated problems are among those that may be answered during consultations.
Morningside and Manhattanville IRB Consultation Service:
Morningside and Manhattanville consultations continue to be offered remotely via Microsoft Teams.
To request a consultation, please email [email protected].
CUIMC IRB Consultation Service (Walk-In Hours) - resuming Thursday, November 6, 2025:
For CUIMC studies that have not yet been submitted, weekly walk-in consultations are now available in-person. These sessions are designed to assist investigators in the early stages of protocol development by providing one-on-one guidance on IRB requirements and submission readiness. In addition, once a month the Reliance Team and the Compliance Oversight Team will be available at the session to provide guidance on reliance and compliance-related matters.
- Location: Hammer Health Sciences Building, Room 314
- Time: Thursdays, 1pm-2pm
Researchers can access the full schedule to non-remote consultations here. No reservations are necessary.
Questions about protocols that have already been assigned to an IRB team should be directed to the HRPO Staff supporting that team. The name of the IRB reviewing your protocol is listed in Rascal on the Protocol Overview Page, next to “Assigned” [IRB 1 – IRB 2 – IRB 3 – IRB 4 – IRB 5 – IRB Exp – Admin – Fac. Review]. Please refer to the HRPO Staff Directory to find the contact information for each IRB team.
Legal requirements to protect human subjects apply to a much broader range of research than many investigators realize. In addition to covering traditional biomedical studies, legal obligations to protect human subjects also apply, for example, to research that uses:
- Human subjects to test devices, products or materials that have been developed through research or to evaluate environmental alterations, for example, habitat modifications.
- Data collected through intervention or interaction with individuals. Intervention includes not only physical procedures (like drawing blood), but also manipulation of a subject's environment and some observations.
- Private information that can be readily identified with individuals, even if the information was not collected specifically for the study in question. Examples include student records and medical records.
- Bodily materials such as cells, blood or urine, tissues, organs, hair, and nail clippings even if you did not collect these materials. (Such research may be considered exempt if materials are not personally identifiable and if the materials were collected prior to the initiation of the research project.)
- Studies conducted to gain generalizable knowledge about categories or classes of subjects such as employees, students, and/or patients. This includes a doctoral dissertation and a master's thesis.
HRPO has developed an IRB guidebook and protocol templates to be used to facilitate the completion of the Rascal application. The links to the guidebook and each protocol template are available on the table below:
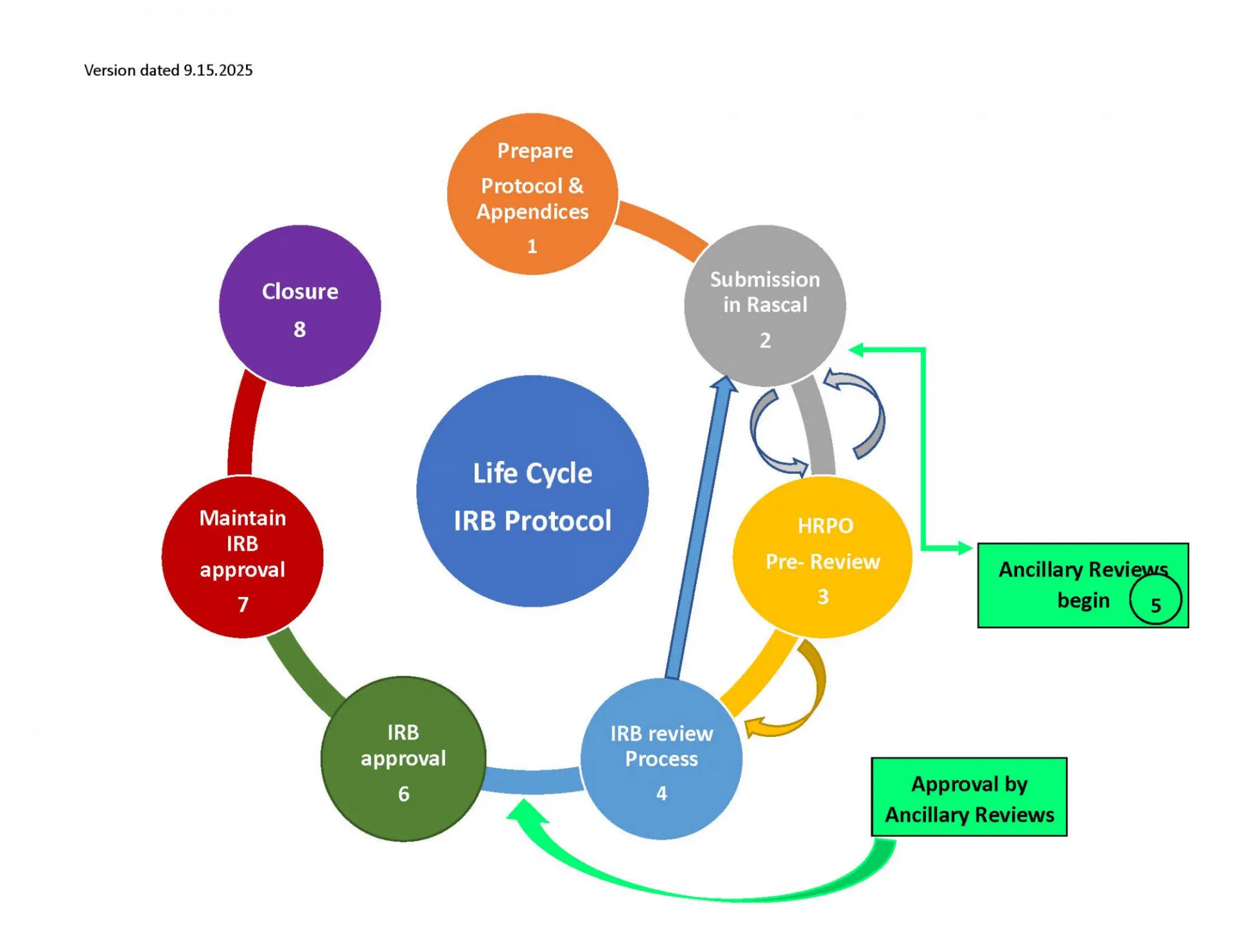
All non-exempt human subjects research protocols require one of the following levels of IRB review.
- Convened IRB Review (also known as Full Board Review): Review at a convened meeting where a valid quorum of IRB members is present. Protocols involving greater than minimal risk or otherwise not eligible to be reviewed by one of the regulatory expedited categories, or by Chair Concurrence or by Limited IRB Review, will be reviewed by the convened IRB.
- Expedited IRB Review: Review by the IRB Chair or an experienced member of the IRB. Protocols involving no more than minimal risk and eligible to be reviewed under one of the expedited review categories defined by HHS.
Limited IRB review: Review by the IRB Chair or designee (hybrid exempt-expedited process). This level of review is for research that falls within one or more of the following four exemption categories:
- Research that only includes interactions involving educational tests when the information obtained is recorded by the investigator in such a manner that the identity of the human subjects can readily be ascertained, directly or through identifiers linked to the subjects [45 CFR 46.104(d)(2)(iii)],
- Research involving benign behavioral interventions in conjunction with the collection of information from an adult subject through verbal or written responses when the information obtained is recorded by the investigator in such a manner that the identity of the human subjects can readily be ascertained, directly or through identifiers linked to the subjects [45 CFR 46.104(d)(3)(i)(C)]
- Secondary research for which consent is not required, [45 CFR 46.104(d)(7)], and 45 CFR 46.104(d)(8). Note that these categories are not applicable at CU.
Under a Limited IRB review, the IRB needs only to confirm that one specific IRB approval criteria is met, which is that there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data [45 CFR 46.111(a)(7)], as opposed to the seven IRB approval criteria that need to be confirmed under Convened IRB review and Expedited IRB review.
- Chair Concurrence: Review by the IRB Chair or designee. This is an alternative to the convened review and is an option that applies only for the review of non-emergency Individual Patient Expanded Access IND requests.
Exempt Research: Human subjects research protocols that fall into several federally-defined "exempt" categories (other than Limited Review exemption categories) are reviewed entirely by HRPO staff. The requirements of the federal regulations for protection of human subjects at 45 CFR 46 do not apply to exempt research.
• Complete all screens in Rascal that are appropriate for a given study before submitting the protocol via Rascal.
• Attach all relevant material, e.g., funding proposal (grant or contract), sponsor protocol, investigator brochure (if the study involves an investigational drug), study instruments [i.e., questionnaire(s) or survey(s)], IND/IDE documentation, non-CU site approvals (e.g., permission to conduct the study).
• If you indicate in the procedures section that the research is cancer-related and you select one of the following five options below*, the protocol will be automatically routed to the Protocol Review and Monitoring Committee queue for review upon login of the Event by HRPO staff.
*Options in Rascal:
o Involves an intervention designed to diagnose, treat, prevent, or provide supportive care to subjects with or at risk of developing a form of cancer.
o Uses specimens or patient information to assess cancer risk, clinical outcomes or response to therapies.
o Utilizes observation or surveillance (no intervention or alteration of patient status).
o Examines outcomes of healthy populations and cancer patients.
o Evaluates the delivery, processes, management, organization or financing of cancer care.
• Verify that information is consistent between documents, e.g., is the number of subjects listed on the data sheet the same as the number in the protocol and/or the consent form?
• Name only one individual as Principal Investigator. This person must be a Columbia faculty of Instruction or Research.
• Have all research personnel complete or update "Personal Information" in Rascal. Use faculty titles, rather than clinical appointments.
Ensure that all personnel have completed the applicable required training(s):
o HIPAA training [TC0019]: required for all research originating from the medical center campus, and for research originating from other campuses that involve PHI.
o HSP (Human Subject Protection) training [TC-0087], including Research with Minors and/or FDA-regulated Research, as applicable: required for all personnel involved in human subjects research. Completion of the HSP basic course is valid for 3 years, after which the HSP refresher training is then required every 3 years.
o S-I (FDA Requirements of Sponsor-Investigators) training [TC0096]: required for Principal Investigators (PI) and Investigators if they are the holder of an IND or IDE.
o Clinical Research Coordinator [TC0098]: required for greater than minimal risk studies or Clinical Research Coordinators or personnel with the following similar roles, such as Regulatory Coordinator, Research Nurse, Data Manager, or Research Assistant.
o Informed Consent in genetic research [TC3700]: required for research coordinators who are obtaining consent for genetic research that is subject to NYS 79-l and for which results are proposed to be returned.
o GCP training [TC3450 or third-party training]: required for personnel on NIH-funded clinical trials. Completion of the GCP basic course is valid for 3 years, after which the GCP refresher training (TC3452) is then required every 3 years .
If non-CU research personnel are involved in the conduct of the research, attach a certificate of completion for equivalent training.
• If requesting an exemption, thoroughly review the exemption categories to ensure that the protocol is eligible for exemption. Consult the IRB staff if necessary. If exemption is requested and the protocol is determined to be ineligible for exemption, it will have to be returned to the investigator to remove the exempt declaration.
• Clearly identify the nature of the data collection, i.e., state whether it is anonymous, de-identified, coded, or non-coded. Explain what mechanisms are in place to protect private, identifiable information.
• Describe in detail how subjects will be recruited (e.g., who will introduce the prospective subject to the study). Attach all advertisements, recruitment letters or other materials that will be used for the recruitment of subjects (e.g., videos, scripts for radio ads, etc.).
• Construct and/or attach all applicable consent documents, (e.g., consent form(s), parental permission form, assent, information sheet), or provide a justification if requesting a waiver of consent in accordance with 45 CFR 46.116(f).
• Include details of appropriate additional protections if subjects may be considered a vulnerable population, e.g., how capacity to consent will be determined, what procedures will be implemented to avoid coercion or undue influence factors.
• Answer all questions on the Protocol-Specific Conflict of Interest form before "signing" the form.
• Attach a cover letter if the protocol includes any factors that may not be self-evident (e.g., eligibility for review per the terms of a cooperative amendment, collaborative relationships, unique funding arrangements, issues that may arise during the review that have been resolved by a specific CU IRB during the review of a similar protocol, etc.)
The checklists below are used by HRPO staff during the administrative pre-review of new protocols, renewals, and modifications.
New Protocol – Rascal Pre-Review Checklist
Modification – Rascal Pre-Review Checklist
Renewal – Rascal Pre-Review Checklist
Research involving Prisoners -- Subpart C: Administrative Reviewer Checklist
Note that the Prisoner Advocate (IRB Member) will complete the following checklist upon review of Research Involving Prisoners (45 CFR 46 Subpart C)-- Prisoner Advocate Checklist: Research Involving Prisoners
This checklist was created by the Department of Pediatrics to facilitate the Department's review of human subjects research and was updated February 12, 2025.
In the Rascal IRB module, “Events” are types of submissions, specifically Protocols, Modifications, Renewals, Annual/Progress Reports, Unanticipated Problems (reports of), and Closures.
• Follow the link in the return correspondence for a list of tasks that should be addressed prior to your next submission.
• Respond via correspondence; be sure to address all questions and concerns as articulated in the Tasks list.
• In lieu of issuing a correspondence, you may download the spreadsheet with the list of tasks and add a column documenting your response to each task. The spreadsheet with your responses should be attached as an External Document, which will show in the Print menu, prior to your resubmission.
• Revise the protocol, consent forms, or other documents as necessary, and attach them as External Documents. A tracked changes version of any revised documents, regardless of whether a change is requested by the IRB or by the study team, should be attached. In addition, clean versions should be attached in pdf format to allow stamping of subject-facing documents.
• Reattach all consent forms built in Rascal that were detached for editing.
• Mark all tasks as completed, as relevant. For any researcher tasks that have not been completed, do not mark the task as “completed.” Instead, provide correspondence addressing why the task has not been addressed or why, in the study team’s opinion, the researcher task is not applicable.
• Archive any superseded or obsolete attached documents.
• If a HIPAA form needs to be updated, first save the current approved form before detaching it, and attach it as a document in the print menu for record purposes. Attach and submit the new updated HIPAA form
• Remember to Notify Approvers to approve and, when all have approved, resubmit the Event.
For additional information on how to review and respond to IRB correspondences, you may review the “New Rascal 2.1 updates” tutorial available in Rascal under the Human Subjects module.
Please visit the webpage, "Maintaining IRB Approval," for more information.

Consent Form Resources
- Information communicated to Researchers on 8/28/2023 following emails from the Columbia University PayCard Team New payment card program for compensation of research subjects
- Notification of Important New Information Letter for Research Participants Letter for Research Participants
- Consent Form Requirements for Studies Linked in Epic
- Addendum to the Informed Consent Document
- Additional Language for the Informed Consent Document
- Additional Language for the Informed Consent Document -Spanish
- Informational Letter for Past Participants for Studies Linked in Epic
- Informational Letter for Past Participants for Studies Linked in Epic - Spanish
Changes to the Consent Process and Forms
The U.S. Department of Health and Human Services and fifteen other Federal Departments and Agencies have issued final revisions to the Federal Policy for the Protection of Human Subjects (the Common Rule). The general compliance date for the Revised Common Rule is January 21, 2019.
The revised Common Rule (2018 requirements) includes changes to the General Requirements for informed consent that are intended to promote autonomy. These changes are described in greater detail below. Accordingly, all non-exempt new protocols submitted for review and approval on or after January 21, 2019 must include the new consent requirements. In particular, the description of the informed consent process on the Recruitment and Consent page in Rascal should consider the new additional General Requirements for informed consent (See section A below) and the consent forms/information sheets submitted with new protocols for approval on or after January 21, 2019 must include the new and basic elements of consent for consideration by the IRB (See section B).
We strongly encourage all consent documents for new protocols submitted before January 21, 2019 to include these elements to facilitate review and approval of your protocol in compliance with the 2018 requirements in the event the protocol is approved on or after January 21, 2019.
A. In addition to the existing general requirements for informed consent, the following requirements have been added:
- Subsection 46.116(a)(4) The prospective subject or the legally authorized representative must be provided with the information that a reasonable person would want to have in order to make an informed decision about whether to participate, and an opportunity to discuss that information.
- Subsection 46.116(a)(5)(i) Informed consent must begin with a concise and focused presentation of the key information that is most likely to assist a prospective subject or legally authorized representative in understanding the reasons why one might or might not want to participate in the research. This part of the informed consent must be organized and presented in a way that facilitates comprehension.
- Subsection 46.116(a)(5)(ii) Informed consent as a whole must present information in sufficient detail relating to the research, and must be organized and presented in a way that does not merely provide lists of isolated facts, but rather facilitates the prospective subject's or legally authorized representative's understanding of the reasons why one might or might not want to participate.
and, when a short form consent process is used - Subsection 46.117(b)(2) A short form written informed consent form stating that the required elements of informed consent required by 45 CFR 46.116 have been presented orally to the subject or the subject's legally authorized representative and that the key information required by 45 CFR 46.116(a)(5)(i) was presented first to the subject, before other information, if any, was provided.
B. In addition to the existing elements of informed consent, the following must be included in the consent form/information sheet, if applicable:
1. To satisfy the requirements of the above-listed subsection 46.116(a)(5)(i), the consent form should include a new section entitled “Key information about this research” that provides a concise summary of the research study.
For Rascal generated consent forms, the section should be placed in the “General Information” section.
For non-Rascal generated consent forms, the section should be placed after the study title and PI contact information.
See additional instructions on what to include in the “Key information about this research” section below.
2. In addition to the existing basic required elements of consent, the following element (45 CFR 46.116(b)(9)) has been added:
One of the following statements about any research that involves the collection of identifiable private information or identifiable biospecimens:
- A statement that identifiers might be removed from the identifiable private information or identifiable biospecimens and that, after such removal, the information or biospecimens could be used for future research studies or distributed to another investigator for future research studies without additional informed consent from the subject or the legally authorized representative, if this might be a possibility; or
- A statement that the subject’s information or biospecimens collected as part of the research, even if identifiers are removed, will not be used or distributed for future research studies.
3. In addition to the existing additional elements of consent, the following elements should be added as applicable (45 CFR 46.116(c)(7) - (9)):
- A statement that the subject’s biospecimens (even if identifiers are removed) may be used for commercial profit and whether the subject will or will not share in this commercial profit;
- A statement regarding whether clinically relevant research results, including individual research results, will be disclosed to subjects, and if so, under what conditions; and
- For research involving biospecimens, whether the research will (if known) or might include whole genome sequencing (i.e., sequencing of a human germline or somatic specimen with the intent to generate the genome or exome sequence of that specimen).
Instructions to complete "Key Information about this research study"
This section of the consent should include a concise summary of the key information that a reasonable person would want to know, in order to help potential research subjects understand why they might or might not want to participate. It is expected that this initial presentation of the key information will be relatively short and should be limited to no more than 1 or 2 pages for consent forms that includes more than 20 pages. For shorter consent forms, this summary should be less than 1 page. Straightforward consent (and assent) forms for non-complex protocols (e.g., that do not contain a complicated methodology) and that are 5 pages or less in length would generally not require a summary of key information. These consent forms are to be presented and organized in a way that facilitates the prospective subject's or legally authorized representative's understanding of the reasons why one might or might not want to participate
Some of the information provided in this section may not need to be repeated in the subsequent sections and reference to additional information in other sections can be made. For some studies, it might be possible to incorporate all required elements into this section, as long as the presentation of that information remains concise and focused. For example, if the study poses no or limited risk and those risks are described in this section, they do not have to be repeated in the RISKS section of the consent form. However, studies with extensive risk profiles for example, should elaborate upon the risks in the RISKS section of the consent form.
This summary should include the most pertinent fact-specific information about this study and should address the following items:
- That consent is being sought for research and that participation is voluntary
- A brief summary of the purposes of the research
- A brief description of the main study procedures (for example, tasks the subject will have to complete, activities the subject will have to avoid). [When writing this section, consider providing responses to the following questions: What are the main reasons a subject will want to join this study? What are the main reasons a subject will not want to join this study? Describe the main things the study will require of the subject (for example, study procedures, tasks the subject will have to complete, activities the subject will have to avoid)]
- Duration of participation
- The most likely risks or discomforts to the prospective subject; [for clinical trials, this section should not list all the risks but identify the risks that would be of highest significance to the subject population, similar to the information that a doctor might deliver in the clinical context in telling a patient how sick the chemotherapy drugs will make them, but with a particular emphasis on how those risks are changed by participating in the study.]
- The most likely benefit(s), if any
- Appropriate alternative procedures or courses of treatment, if any that might be advantageous to the potential subject. [When writing this section, consider providing responses to the following questions: What impact will participating in this research have on the subject outside of the research? For example, will it reduce options for standard treatments? How will the subjects’ experience in this study differ from treatment outside of the study? In what ways is this research novel?]
Example template language:
The purpose of this study is…
The procedures in this study are … We will ask you to do… The time frame for the study is… The detailed information in sections XX and XX below gives full instructions, and the full schedule for visits is in section XX.
The reasons you might not want to participate in this study are… Risks include… All of the known risks are listed in section XX.
The reasons you might want to participate in this study are… For a complete description of possible benefits, please see Section XX below.
Instead of volunteering for this study, you have other choices… You can choose to get treatment from your doctor or clinic, or from [name/clinic]. You can find out if there are other studies that may be of more interest to you [add how to find out]. If you decide to take part in the study, it should be because you really want to volunteer. You can choose to withdraw at any time during the study. If you choose not to volunteer you will not lose any services, benefits, or rights you would normally have.
If you are interested in learning more about this study, please read the details below.
Benefit to using the RASCAL Consent Form Builder: As soon as the status of the protocol changes to "approved", the consent form will receive the IRB approval stamp electronically and can immediately be printed by the researcher.
- Create a generic form once; copy and edit as necessary for each study.
- Utilize the three ways to insert text: a) select sample text; b) copy/paste from another document; c) type.
- Typing or copying text: remember that the Consent Form Builder is a web program and recognizes only basic formatting.
- Inserting text from a sample consent: All text related to one element of consent or topic (e.g., Procedures, Risks, etc.) can be copied into their respective field in the RASCAL Consent Form Builder.
- Bolding of headings: RASCAL automatically bolds the headings of the elements of consent (e.g., Purpose and Procedures, Risks, Benefits, etc.), when the consent is created utilizing the Consent Builder.
- Additional headings of a given element of consent or topic (e.g., Risks of Pregnancy under Risks) should be entered in the sub-heading field in RASCAL.
- Otherwise, inserting an entire consent form into a field in the RASCAL Consent Form Builder, go to Layout and enter a "0" into all fields other than the one that contains the consent form. This process will not print the headings in bold. This process is not recommended by the IRB as it would decrease comprehension of the consent document.
- Be familiar with the required elements of informed consent, and include the additional elements when appropriate.
- Review the "Helpful Consent Form Information" available from the Consent Form menu.
- Read the general instructions in each section of the consent builder. These will guide you to submit a consent form to the IRB more successfully.
- Ensure that information in the consent form is consistent with that in the protocol and all other study-related documents.
- Maintain a 6th-8th grade language level throughout the form. See the "Tips for Increasing Comprehension of Informed Consent Documents" below.
- Maintain the same voice (second person; e.g., "you") throughout the consent form.
- Select signature lines cautiously, i.e., include either "study participant" or "study subject" but not both. Remove any unnecessary or inappropriate signature lines.
- Minimally, a consent document that will be signed should have the following lines for signature and date: 1) Signature of the Subject or Participant; 2) Signature of the Person Obtaining Informed Consent; and 3) Investigator's signature.
- The Signature of the Subject or Participant may not be appropriate for all assent forms.
- After attaching the consent form to a protocol and before submitting it to the IRB, print it out and read it through to evaluate layout, appearance, flow of text, language level and accuracy. Ensure that all required elements of consent are included or justify the removal of any element of consent in the Subjects page of the protocol.
- If a consent form includes a chart or table, move the chart or table into a separate document that will be used as an addendum to the consent. Attach the addendum in RASCAL and explain in a cover letter that the attachment should be considered as part of the consent form.
- If an approved consent document needs to be translated into another language, the investigator can grant access of the consent document within RASCAL to the individual who will conduct the translation. For example, if a consent form needs to be translated into Spanish, the investigator can grant access to an individual in the Hispanic Research & Recruitment Center (HRRC). Alternatively, one can attach a scanned copy of the translated form(s) stamped by the HRRC.
- If similar text is used or required by several investigators or throughout a department, provide the sample text to the IRB. The IRB will consider such requests and revise sample text in the Consent Builder accordingly.
- Contact the RASCAL help-line if technical assistance is required (whenever you are unable to perform any function in the Consent Form Builder or manipulate or edit any text).
- Contact the IRB office if questions regarding institutional policy and/or federal regulations arise during construction of the consent form.
All informed consent documents not created in RASCAL should be printed on CUMC letterhead or include information identifying the institution and research team or center. One inch margins are to be used on either side and on the bottom with 1.5 inches at the top. The type size to be used should be 12 point. Number all pages and use a header beginning with the second page of the document.
The document should be written at the eighth grade reading level. Suggestions for meeting an eighth grade reading level include:
- Use one or two syllable words whenever possible
- Write short sentences and paragraphs
- Define all medical or technical terms in lay language
- Organize information in sections with clear headings
- Print all headings in bold
- Use spacing to emphasize important concepts
- Avoid contractions such as don't
Address the consent document to the reader by using the active voice and the word "you" throughout, i.e. "You are being asked to take part in a research study . . ."
Detailed information on do's and don'ts, complex drug dosing, and concomitant medications should not be included in a consent form. This information could be in a separate information sheet for subjects.
Providing information in a question and answer format is one method of increasing comprehension. This format is particularly useful when the study procedures are complex and the target subject population has a below average education or reading level.
Include version number and/or version date on each version of a consent document.
- The Consent Process: The informed consent process involves an ongoing dialogue between the subject and the investigator that continues until the subject's participation is complete. People who agree to participate in research must first understand and agree to volunteer based on the information provided to them by the researcher. That information must include details about the research, how long it will last, how many visits it will take, and all the risks and benefits. In giving informed consent, subjects may not waive or appear to waive any of their legal rights, or release or appear to release the investigator, the sponsor, the institution or agents thereof from liability for negligence. Upon meeting with a potential subject, an investigator must first determine whether he or she is capable of giving consent. If so, the consent process begins by discussing the purpose, risks, and benefits of participation, following which the subject must be allowed sufficient time to consider all options and ask questions. The investigator must ensure that information is conveyed in a manner that the subject can easily understand and make sure the subject has understood the information provided before obtaining the subject's voluntary informed consent to participate in the research protocol. In this ongoing process of consent, Investigators must maintain the dialogue with subjects by answering additional questions, by providing additional information as it becomes available, and by reaffirming the subject's willingness to participate throughout the study.
- Vulnerable Populations: There may be some subjects who require special considerations, such as children, prisoners, cognitively impaired individuals, economically/educationally disadvantaged individuals and others. Consideration for the protection of these "vulnerable populations" is integral to IRB review.
- Consent Form: A written document that embodies the elements of informed consent required by 45CFR46.116. This form may be signed by (or read to) the subject or the subject's legally authorized representative. The investigator must give either the subject or the representative adequate opportunity to read the consent form and ask questions about the research before the subject or the representative signs the consent form or provides verbal consent to participate in the research protocol. Note:Only consent forms that have been previously submitted to and approved by the IRB may be used.
- Assent: Agreement by an individual not able to give legally valid informed consent (e.g., a child or cognitively impaired person) to participate in research.
- Parental Permission: A form signed by one or both parents agreeing to enroll their child in research. The parental permission must include all the elements of consent and a copy must be provided to the parent(s).
- Information Sheet: Document provided to research subject that contains the elements of informed consent but does not require a signature. Used either in exempt research, (in which informed consent is not required but may be desirable) or when written documentation of informed consent has been waived by the IRB in accordance with 45CFR 46.117(c).
- Legally Authorized Representative: An individual authorized under applicable law to consent on behalf of a prospective subject to the subject's participation in the procedure(s) involved in the research. In New York State, an LAR is a parent of a minor or a court appointed guardian.
Pre-2018 Requirements (applicable to protocols initially approved by the IRB prior January 21, 2019 (identified in Rascal as "Legacy Protocol"))
General Requirements for Informed Consent
Waiver of Elements of Informed Consent
Waiver of Documentation of Informed Consent
2018 Requirements (applicable to all protocols initially approved by the IRB on and after January 21, 2019)
General Requirements for Informed Consent 2018 Requirements
Waiver of Documentation of Informed Consent 2018 Requirements
Waiver or Alteration of the Elements of Consent_ 2018 Requirements
General
- Consent Form Builder Sample Language
- Information Sheet: Consent and HIPAA Authorization
- Incidental Finding Consent Template Language
- Radiation Risk for Studies Using Ionizing Radiation
- Model MRI Risk Information for IRB Consent Forms
- Addendum to Consent Document Template
Minimal Risk Consent Form Templates
- Minimal Risk Consent Form Template (no recording)
- Minimal Risk Consent Form Template (studies involving audio/video recording)
- Standard Format for Social/Behavioral Science Consent Documents
Assent Form/Script Templates
- Assent Form (for children ages 7-11)
- Assent Form (for children ages 12-17)
- Assent Form (for children ages 7-12) - Research Study Involving exome/genome sequencing
For use when the primary focus of a study is whole exome (WES) or whole genome sequencing (WGS):
Consent Form Addenda regarding CUMC IRB Office move: